В.Г. Антонов, В.К. Козлов Российская Военно-медицинская академия МО РФ; Санкт Петербургская медицинская академия последипломного образования МЗ РФ Патогенез онкологических заболеваний.
В.Г. Антонов, В.К. Козлов Российская Военно-медицинская академия МО РФ; Санкт Петербургская медицинская академия последипломного образования МЗ РФ Патогенез онкологических заболеваний.
Цитоплазматические и молекулярногенетические механизмы иммунной резистентности малигнизированных клеток В обзоре рассматривается роль окислительно-восстановительных реакций, белков теплового шока, протеосомного комплекса, каспаз как цитоплазматических основ иммунной резистентности опухолевых клеток. Анализируются механизмы генетической регуляции активности этих факторов, прото и антионкогенов, а также участие цитокинов в этих процессах. (Цитокины и воспаление. 2004. Т. 3, № 2. С. 23–33.)
В первой части проблемной статьи доказывалось, что основными молекулярными механизмами устойчивости малигнизированных клеток к эффектам иммунной системы являются избирательная десенситизация рецепторов мембран, экранирование антигенных детерминант специфическими антителами, белками теплового шока и другими молекулами, Fas зависимый и Fas независимый варианты инактивации иммуноцитов. Итогом действия данных механизмов оказывается ограничение, но не отмена воздействия регуляторных и эффекторных молекул и клеток иммунной системы. Злокачественно трансформированные клетки сохраняют также способность селективно взаимодействовать с цитокинами, включая семейство белков фактора некроза опухоли. В биологической активности цитокинов при этом превалирует регуляторная составляющая их активности, что может только способствовать развитию малигнизированных клеток.
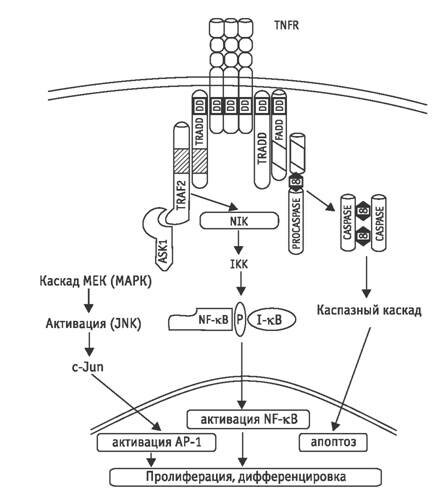
Цитоплазматические механизмы иммунной толерантности малигнизированных клеток Действие цитокинов на клетки, включая злокачественно трансформированные, приводит к индукции гибели клетки путем апоптоза или некроза, стимуляции процессов клеточного деления с увеличением клеточной массы, инициации процессов дифференцировки и появлению функционально дееспособных клеток [3]. Составляющие регулярного воздействия фактора некроза опухоли иллюстрирует рис. 1. Инициация апоптотического и антипролиферативного сигналов осуществляется наряду с индукцией деления и дифференцировки клеток. Нормальные клетки, способные к взаимодействию с цитокинами, но не готовые к делению, дифференцировке и другим физиологическим реакциям, элиминируются путем апоптоза. В цитоплазме малигнизированных клеток выявляется практически весь спектр молекул, способных обрабатывать регуляторные сигналы цитокинов и включать определенные клеточные функции. Подобная детерминированность молекулярного представительства в цитоплазме обусловлена эволюционно сложившимся использованием одних и тех же молекул как в сигнальных, так и в эфферентных системах [12]. Приоритет в выборе сигнального пути и сопряженной с ним клеточной функции определяется цитоплазматическими механизмами селекции регуляторного воздействия цитокинов на клеточные процессы. В малигнизированной клетке именно эта система структурной организации сигнальных потоков в цитоплазме обеспечивает нейтрализацию апоптотического и антипролиферативного компонентов регуляторного воздействия цитокинов, обусловленную системами глутатиона, тиоредоксина, протеолитического контроля клеточных функций и белков теплового шока. Ниже рассматриваются биохимические закономерности функционирования цитоплазматического компартмента обеспечения иммунной резистентности опухолевых клеток. «Редокс-модуляция» сигнальных молекул. Для окислительно-восстановительной («редокс») модуляции активности сигнальных молекул в опухолевых клетках характерны те же закономерности функционирования, что и для нормоцитов. Возможность обратимой окислительно-восстановительной модуляции активности молекул, осуществляющих сигнальные функции, определяется наличием в их структуре тиоловых групп. Регуляция осуществляется модулирующими соединениями в соответствии с формулой SH SH ?модулирующее соединение? S S. Физиологическими модуляторами активности регуляторных молекул во всех клеточных компартментах и в ближайшем микроокружении клеток выступают окисленная и восстановленная формы глутатиона (GSSG и GSH соответственно), белок тиоредоксин, органические и неорганические радикалы, оксид азота (NO) [5, 15, 48]. «Редокс-модификация» тиоловых групп приводит к кардинальному изменению активности сигнальных молекул.
Рис. 1. Схема сигнальных потоков в клетке при воздействии TNF? (по [3] с изменениями)
Цитокины, хемокины, ростовые факторы, пептидные гормоны, молекулы межклеточных взаимодействий и молекулы внеклеточного матрикса, а также большинство рецепторов плазмалеммы повышают свою функциональную активность при окислении тиоловых групп. Напротив, при восстановлении тиоловых групп функциональная активность этих молекул уменьшается или утрачивается. В цитоплазме складывается более сложная зависимость между активностью молекул сигнального звена и химическим состоянием тиоловых групп в домене или доменах белков, непосредственно обеспечивающих передачу регуляторного сигнала. Для функционально активного состояния протеинкиназ необходимо наличие восстановленных тиоловых групп в структуре каталитического домена и, возможно, присутствие дисульфидной сшивки в структуре другого домена [4, 15]. Фосфатазы, осуществляющие негативную регуляцию, в функционально активной форме имеют в молекуле дисульфидную сшивку, которая обеспечивает их активность. Активные формы протеинкиназ, фосфатаз и другие сигнальные молекулы цитоплазмы необходимы для упорядочивания и сортировки регуляторных сигналов. Необходимый статус тиоловых групп в сигнальных молекулах цитоплазмы обеспечивается сродством тиола к модулирующему его активность соединению, стерической доступностью тиоловой группы и концентрацией модулирующего агента. Для опухолевых клеток по сравнению с нормоцитами характерна повышенная способность к восстановлению тиоловых групп [35]. Опухолевые клетки сохраняют лишь минимальный уровень молекул, активность которых требует наличия в их структуре дисульфидной сшивки. Химические соединения, которые осуществляют окислительную модификацию тиоловых групп, также продуцируются опухолевой клеткой в значительно меньших количествах, чем нормоцитами. В контексте вопроса о цитоплазматических механизмах иммунной резистентности опухолевых клеток, которые обеспечиваются негативной модуляцией апоптотического и антипролиферативного сигналов цитокинов, возможна реализация следующих механизмов. Восстановленное состояние тиоловых групп в структуре молекул сигнального звена цитоплазмы способствует прохождению сигналов различного характера. Однако восстановленные тиоловые группы связывают свободные радикалы, нейтрализуют их и препятствуют модификации их субстратов.
В итоге ограничивается проапоптотическое влияние цитокинов семейства фактора некроза опухоли, а апоптоз не реализуется [50, 55]. Восстановленное состояние тиолов в белках митохондриальной поры обеспечивает так же депонирование проапоптотических факторов митохондрий [46, 58, 60], что препятствует участию соответствующих цитокинов в мобилизации митохондриальных факторов апоптоза. Так, наличие в клетке восстановленного глутатиона и активированное состояние системы тиоредоксина необходимы для обеспечения постоянной готовности энзимов к репарации. Протеолитический контроль регуляторного воздействия цитокинов. Механизмы протеолитического контроля высокоэффективны в клетке благодаря их структурной простоте и энергетической экономичности. Реакции протеолиза необратимы и одноступенчаты, поэтому их результат всегда однозначен. Протеолитический контроль, являясь относительно самостоятельным регуляторным механизмом, имеет точки сопряжения с эволюционно более поздними и динамичными сигнальными механизмами. Однако в нормоцитах именно этот механизм предопределяет итоговый ответ клетки на внешнее воздействие, включая цитокины и другие гуморальные регуляторные молекулы иммунной системы. В злокачественно трансформированных клетках возможности протеолиза ограничены, он осуществляется лишь некоторыми каспазами, а активность протеосомы и действие лизосомальных ферментов направлены лишь на определенные субстраты. Избирательность в активности различного рода систем протеолиза резко снижает эффективность ответа малигнизированной клетки на проапоптотический и антипролиферативный сигналы цитокинов. В этой связи недостаточная активность системы протеолиза в злокачественно трансформированных клетках может рассматриваться как цитоплазматический механизм формирования иммунной толерантности. Таким образом, депрессия антипролиферативной и проапоптотической составляющих действия цитокинов на малигнизированные клетки достигается уменьшением выраженности и интенсивности реакций протеолиза, которые в свою очередь обеспечиваются активностью протеосомы и/или соответствующих каспаз. Формирование иммунной резистентности трансформированных клеток с участием протеосомы. Протеосома—это протеолитический мультисубъединичный комплекс, состоящий из каталитически активного ядра и регуляторных субкомплексов [13, 30, 44]. Гетерогенность субъединиц протеосомы является структурной особенностью и предопределяет разнообразие форм протеолитического комплекса, а также его широкую субстратную специфичность [51, 57, 58]. Протеосома осуществляет АТФ и убиквитин-зависимый протеолиз онкобелков и белков-супрессоров опухолей [32, 39], факторов транскрипции [39], регуляторов клеточного цикла [38, 62], ферментов метаболизма ДНК [45, 47]. Следовательно, именно благодаря активности протеосомы в клетке формируется пул антигенных пептидов, которые могут играть роль опухоль-ассоциированных антигенов и экспрессируются совместно с молекулами главного комплекса гистосовместимости I класса на клеточной поверхности злокачественно трансформированной клетки [22, 29, 61]. Представление этих антигенов дендритными и другими клетками позволяет специфическим цитотоксическим Т лимфоцитам распознавать и уничтожать клетки, экспрессирующие полипептиды, которые не свойственны нормоцитам [1, 22]. В настоящее время накоплено достаточное количество данных, свидетельствующих о прямом участии протеосомы в системах контроля клеточного деления, а также об опосредованном участии протеосомы в иммунном контроле состояния клетки. Проонкогенные изменения активности протеосомы, позволяющие опухолевой клетке преодолевать систему иммунного контроля, идентифицированы в таких субстратах протеолиза, как антигенные полипептиды, структурные онкобелки, опухолевые супрессорные белки. Антигенные полипептиды. Любые синтезированные в клетке белки подвергаются деградации протеосомой по АТФ и у биквитин зависимому путям.
В результате гидролиза образуются полипептиды, которые включают от 5 до 24 аминокислот. Часть этих полипептидов является иммунологически значимыми эпитопами опухолевых антигенов. В цитозоле состоящие из 8–11 аминокислот полипептиды соединяются с молекулой ТАР (transporter associated with antigen presentation) и транспортируются в эндоплазматический ретикулум, где образуют комплекс с молекулами МНС I класса, а затем выносятся на поверхность клет ки. Представление этих антигенов дает возможность иммунной системе распознавать и разрушать клетки, экспрессирующие необычные полипептиды [22, 39]. Из внеклеточных биорегуляторов в контроле процесса образования антигенных полипептидов участвует IFN?, воздействие которого на клетки повышает экспрессию отдельных субъединиц протеосомы и возникновение ее новых форм, способных увеличивать количество и спектр презентируемых пептидов [58, 59]. Тип протеосомы, индуцируемый IFN?, назван «иммунопротеосомой». Канцерогены и коканцерогены, атакже ряд белков, экспрессируемых в клетке при поражении онкогенными вирусами (EBV, HTLV 1, HBV, HCV, HSV), вероятно, способны негативно влиять на образование иммунопротеосомы и продукцию антигенных полипептидов. Снижение способности протеосомы к генерации антигенных полипептидов предопределяет антигенную невыразительность трансформирующихся и малигнизированных клеток и обеспечивает их ускользание от системы иммунного надзора. Структурные онкобелки и опухолевые супрессорные белки. Деградация р53—ключевого внутриклеточного белка-супрессора злокачественной трансформации клетки—происходит по убиквитин-зависимому протеосомному пути. Низкий уровень р53 отмечен при злокачественной трансформации, обусловленной онковирусами и коканцерогенами. Интенсивность образования мРНК данного белка и скорость его синтеза соответствуют значениям нормальных клеток [43]. В малигнизированных клетках снижение количества р53 является следствием ускоренной деградации этого белка, в том числе в протеосоме, при избирательном стимулирующем действии коканцерогенов. В случае вирусного коканцерогенеза возрастает количество ассоциированного с р53 белка Е6 АР, что инициирует убиквитинирование и ускоренную деградацию р53 [24]. Подобные варианты изменения количества белка р53 ограничивают возможности реализации апоптотических сигналов по р53 зависимому пути. Проапоптотическая активность цитокинов также может быть преодолена в условиях избытка белка с myc [8], уровень которого в нормальных клетках обычно очень низок, а в опухолевых — высок. Формирование иммунной резистентности малигнизированных клеток с участием каспаз. Каспазы являются эволюционно консервативными цистеиновыми протеазами, распознающими в белках аминокислотную последовательность Р4?Р3?Р2?Р1, в которой позиции Р1 и Р3 занимают остатки аспарагиновой и глутаминовой кислот [49, 61]. По уровню ферментативной активности каспазы подразделяют на низкоактивные прокаспазы и высокоактивные собственно каспазы. Каспазы образуются в результате ограниченного протеолиза и олигомеризации соответствующих прокаспаз, чья активность составляет несколько процентов от активности каспазы. По характеру преобладающего участия в физиологических процессах выделяют несколько групп каспаз. Активаторы цитокинов—каспазы 1, 4, 5, 13—осуществляют процессинг цитокинов. Инициирующие (учреждающие)—каспазы 2, 8, 9, 10—интерпретируют регуляторный сигнал от внеклеточных (каспазы 8, 10) и внутриклеточных (каспазы 2, 9) биорегуляторов тех или иных физиологических процессов. Эффекторные—каспазы 3, 6, 7—осуществляют реализацию регуляторного сигнала в соответствующем физиологическом процессе. Специализация инициирующих каспаз обеспечивается структурой их специализированных доменов. Каспазы 8 и 10 имеют полипептидный участок DED (a death effector domain), который участвует в гидрофобных взаимодействиях. Каспазы 2 и 9 имеют полипептидный участок, обозначаемый как CARD (caspase activation and recruitment domain). Этот участок обеспечивает электростатические взаимодействия каспаз. Воздействие регуляторных молекул иммунной системы, опосредованное активацией рецептора, реализуется посредством трансформации доменов DED или CARD соответствующих прокаспаз и мобилизует активность инициирующего фермента, который в свою очередь запускает соответствующую эффекторную каспазу [23, 54]. В нормальных клетках стационарная активность каспаз необходима для обеспечения их жизнедеятельности. Например, в секретирующих ростовые факторы нормоцитах каспазы 1, 4, 5, 13 завершают процесс образования активных форм этих факторов [48]. Активация транскрипционного фактора NF ?B, необходимая для индукции физиологического ответа клетки, также возможна по зависимому от активности каспаз механизму. В этом случае каспазы осуществляют протеолиз ингибиторов NF?B. Деление нормальных клеток обычно происходит при участии каспаз. Каспазы расщепляют белки р21WAF/CIP1 и р27KIP1, которые являются ингибиторами комплексов циклин D Cdk4,6; циклин E Cdk2, циклин A Cdk2 и циклин В Cdс2, предопределяющих возможность прохождения клеткой цикла деления [26]. Миграция нормальных клеток в структуре ткани и их последующая дифференцировка блокируются ингибиторами каспаз [56]. Малигнизированные клетки с присущими им экспрессией ростовых факторов, митотической активностью, инвазивным ростом и другими характерными свойствами имеют практически весь спектр каспаз, выявляемый в нормоцитах. Как и в нормоцитах, в злокачественно трансформированных клетках сохранена способность каспаз к выполнению основных функций. Однако в малигнизированных клетках проявление этих функций каспазами носит избирательный характер: каспазы участвуют во всех присущих им физиологических реакциях за исключением апоптоза.
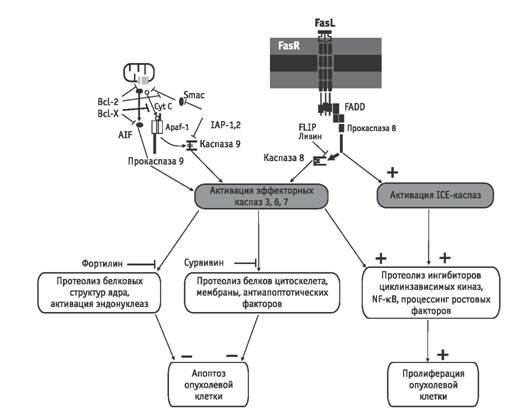
Подобное состояние приводит к тому, что воздействие на малигнизированные клетки регуляторных молекул иммунной системы, обычно инициирующих апоптотическую реакцию каспаз, не вызывает апоптоза, и злокачественно трансформированные клетки не погибают. Напротив, ответной реакцией малигнизированной клетки может быть инициация клеточного цикла, что обусловлено избирательной активацией каспаз при действии цитокина. Подобный характер ответа опухолевых клеток на инициирующее воздействие регуляторных молекул иммунной системы связан с повышенной экспрессией в этих клетках селективных ингибиторов каспаз. Механизмами негативного воздействия ингибиторов на активность каспаз являются подавление образования активированных форм каспаз из прокаспаз, нейтрализация активных форм каспаз, блокирование мобилизации каспаз из цитоплазматических депо. Компартментализация ингибиторов каспаз свойственна как малигнизированным клеткам, так и нормоцитам. В опухолевых клетках избыточные концентрации ингибиторов каспаз определяются в микроокружении цитоплазматических субстратов, протеолиз которых обеспечивает необратимость процесса клеточной гибели. Белковые ингибиторы каспаз, которые связываются с молекулой фермента и/или профермента, составляют семейство IAP (inhibitor of apoptosis). В цитозоле опухолевых клеток повышена экспрессия ингибиторов различной специфичности. Цитозольными ингибиторами каспаз являются ливин и casper (синонимы FLIP/CASH/CLARP/FLAME 1/I FLICE/MRIT/usurpin), которые селективно подавляют передачу проапоптотического сигнала на прокаспазу 8. Прокаспаза 8 (FLICE)—конститутивный протеолитический фермент, необходимый для нормальной жизнедеятельности при уровне ее активности порядка 1–2 % максимально возможной. Прокаспаза 8 активируется протеиновым комплексом TNFL?TNFR1?TRADD?FADD, включающим белок FADD (рис. 1). После взаимодействия с белком FADD происходит аутопроцессинг и димеризация прокаспазы 8 с развитием максимальной активности. Каспаза 8 обеспечивает прирост до неконтролируемого уровня активности ряда других каспаз, что приводит к протеолизу клеточных структур и гибели клетки путем апоптоза. Ингибиторы активности каспазы 8—белки ливин и casper конкурируют с ней за возможность взаимодействия с белком FADD [15, 54], что ограничивает активность фермента, однако в злокачественно трансформированных клетках этот механизм не работает. Кроме способности ингибировать процессинг каспазы 8 белок ливин ингибирует также каспазу 9. Аналогичные функции выполняет и цитозольный белок IL-P 2. Инициаторные каспазы обоих типов способны также ингибировать белок BAR (bifunctional apoptosis regulator), который взаимодействует с прокаспазами 8 и 10, препятствуя их связыванию с белком FADD с последующей активацией соответствующих каспаз. Взаимодействие между белком BAR и антиапоптотическими белками Bcl 2 и Bcl хL защищает последние от и нактивации белком Вax и препятствует освобождению из митохондрий прокаспазы 9 и других факторов [63]. Помимо повышенной экспрессии белков-ингибиторов, инициирующих каспаз 8, 9, 10, в малигнизированных клетках повышено также содержание белков-ингибиторов эффекторных каспаз 3, 6, 7. Эти ингибиторы локализованы преимущественно в области тех субстратов, протеолиз которых каспазами приводит к запуску апоптоза. В опухолевых клетках часто обнаруживают белки с функцией ингибиторов каспаз — сурвивин и фортилин. Высокое содержание сурвивина характерно преимущественно для микротрубочек цитоскелета, а фортилина—для клеточного ядра [16]. Сурвивин и фортилин подавляют активность каспаз 3 и 7, субстратами которых являются актин и связывающие актин с плазмалеммой белки гельзолин и фодрин [18]. Ингибирующее действие сурвивина и фортилина сказывается на процессах фрагментации ядра, образования везикулоподобных выпячиваний мембраны и апоптозных телец. Однако вне зоны своей локализации сурвивин не ограничивает участия ингибируемых каспаз, что реализуется в малигнизированных клетках. Активированная форма каспазы 3 расщепляет белок р21WAF/CIP1, который является ингибитором комплексов циклинов Е и А с Cdk2 и циклина В с Cdc2, определяющих возможность прохождения злокачественно трансформированной клеткой цикла деления [26]. Другой составляющей активности каспазы 3 является стимуляция экспрессии антиапоптотических белков митохондрий семей ства Bcl 2 и цитоплазматических белков IAP1 и IAP2 [25]. Так, каспаза 3, активированная каспазой 9, способна осуществлять ограниченный протеолиз каспазы 9 с отделением от ее молекулы фрагмента, активирующего фактор NF ?B, который в свою очередь инициирует экспрессию белков Bcl 2 и IAP [25, 53, 54]. Развитие апоптоза при действии цитокинов на малигнизированные клетки требует не только активации цитоплазматических каспаз, но и мобилизации проапоптотических факторов митохондрий, включая цитохром С, прокаспазы 2, 3 и 9, белок AIF и другие факторы [60]. Следовательно, одним из действенных механизмов ингибирования апоптоза в злокачественно трансформированных клетках является устойчивая компартментализация этих факторов, обеспечиваемая многочисленными белками семейства Bcl 2. В настоящее время клонировано 16 генов, определяющих их синтез. Однако антиапоптотической активностью обладают Bcl 2, Bcl хL, Bcl w, Boo, Al, Mcl, тогда как Bax, Bak, Bok/Mtd, Bad, Bik/Nbk, Bid, Bik, Bim/Bod, Hrk/DPS, Bcl xS индуцируют развитие апоптоза. В «преуспевающих» малигнизированных клетках экспрессируются антиапоптотические белки этого семейства, но ограничена экспрессия проапоптогенных форм [37]. Белки семейства Всl 2 необходимы для сохранения непроницаемости митохондриальных пор для проапоптотических факторов. Такая конформация белков митохондриальной поры обеспечивается восстановлением ихтиоловых групп. Антиапоптотические белки семейства Всl 2 экранируют тиоловые группы от окисляющих агентов и/или инициируют механизм их восстановления в случае образования дисульфидных сшивок, что обеспечивает минимальный диаметр и непроницаемость митохондриальной поры для проапоптотических факторов [46, 60, 63]. Рис. 2 иллюстрирует функционирование механизма иммунной резистентности в цитоплазматическом компартменте с участием ингибиторов каспаз и антиапоптогенных белков семейства Вcl 2. Формирование иммунной резистентности трансформированных клеток с участием белков теплового шока. Белки теплового шока (HSP) являются древней защитной системой клетки, обеспечивающей выживание клетки в стрессовых условиях. Определенные HSP присутствуют в клетке конститутивно, другие экспрессируются в клетке при воздействии стрессорных факторов, и повышение их концентрации носит транзиторной характер [10]. В разные периоды развития опухоли ее клетки постоянно испытывают различные воздействия со стороны макроорганизма, направленные на ограничение их роста и уничтожение. Подавляющее большинство
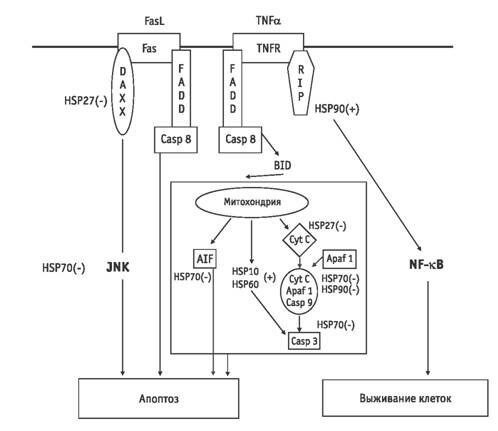
Рис. 2. Негативный контроль активности проапоптотических реакций в цитоплазме посредством специфических ингибиторов активности каспаз и белков семейства Всl
2
малигнизированных клеток при этом гибнет путем апоптоза. Выжившие в первичном очаге злокачественно трансформированные клетки, равно как и клетки метастазов, продолжают развиваться в условиях хронического стресса и селективного давления со стороны нормоцитов, продуцирующих провоспалительные цитокины (TNF?, IL-1, IL-6), и эффекторов врожденного противоопухолевого иммунитета. В этой связи закономерностью становится перманентное повышение уровня индуцибельных HSP, которые обеспечивают защиту опухолевой клетки по отношению к иммунным факторам организма хозяина. Механизм функционирования HSP в сопряжении с плазмалемой и экранирование антигенных детерминант опухолевой клетки был проанализирован в предыдущей публикации [2]. В цитоплазме опухолевых клеток также экспрессируется широкий спектр HSP, обеспечивающих депрессию проапоптотического и антипролиферативных влияний цитокинов. При этом экспрессируемые в опухолевой клетке HSP реализуют негативное воздействие на составляющие как внутреннего (митохондриального), так и внешнего (рецепторного) контуров проапоптотических сигналов. В частности, HSP70 ингибирует процессы выхода цитохрома С и активации каспазы 3 [40], нарушает структуру и снижает активность апоптосомы [19]. HSP70 тормозит апоптоз опухолевых клеток, индуцированный белком AIF [41]. Подобно специфическим игибиторам каспаз, HSP70 обеспечивает защиту опухолевых клеток от TNF индуцируемого апоптоза, не влияя на активацию прокаспазы 3, но экранируя критические субстраты от протеолитической модификации [36]. Кроме HSP70 антиапоптотическими эффектами обладают и другие белки-шапероны. Так, HSP27 осуществляет негативную регуляцию апоптоза, блокируя адапторные белки, ассоциированные с цитоплазматическими участками рецепторов для TNF?, а HSP90 препятствует формированию апоптосомы, взаимодействуя с RIP 1 киназой (receptor interacting protein), что предопределяет выживание клеток при участии NF ?В [55]. HSP70 также может ингибировать активность проапоптотической JNK киназы, ограничивая прохождение сигнала от Fas рецептора. Рис. 3 иллюстрирует точки приложения активности белков теплового шока, обеспечивающих негативную регуляцию внешнего и внутреннего апоптотических каскадов опухолевых клеток. Вклад белков теплового шока в комплекс цитоплазматических механизмов иммунной толерантности не ограничивается их антиапоптогенной активностью. Белки теплового шока HSP70, 90 и 23 определяют также уровень экспрессии стероидных рецепторов и, соответственно, являются посредниками цитопротекторной активности стероидов по отношению к опухолевым клеткам.
Таким образом, биохимические механизмы иммунной резистентности малигнизированных клеток как на цитоплазматическом, так и на мембранном уровнях ее биологической организации представляют собой вторичный фенотипический признак. Так как первичные механизмы формирования иммунной резистентности при этом не затрагиваются, то очевидно, что ингибирование ключевых составляющих биохимических механизмов обеспечения иммунной толерантности опухолевых клеток будет лишь паллиативной мерой. Вероятно, механизмы формирования иммунной резистентности злокачественно трансформированных клеток следует рассматривать в едином контексте с биохимическими проявлениями их метаболизма.
Тот факт, что малигнизированная клетка—это генетически измененная клетка, указывает на связь механизмов формирования ее иммунной резистентности с функциональной активностью продуктов соответствующих дефектных генов.
Рис. 3. Регуляция белками теплового шока процессов апоптоза в опухолевых клетках (по [28]): (+) — активирующее, (–) — ингибирующее воздействие HSP на соответствующий аффинный белок
Молекулярно-генетические аспекты формирования иммунной толерантности злокачественно трансформированных клеток Канцерогенез—это процесс длительного накопления в клетке мутаций и «эпимутаций» про то и антионкогенов [27, 52]. С помощью новых методов контроля экспрессии генов (например, ДНК микрочипов) установлено, что в малигнизированной клетке изменены сотни генов [31, 42, 49]. Последовательность приобретения перерождающейся клеткой генетических дефектов строго не предопределена, но и не абсолютно случайна. Предпочтение в порядке приобретения генетических дефектов обусловлено тем, что в скрытый период злокачественного перерождения трансформированная клетка должна ускользать от системы врожденного противоопухолевого иммунитета. Низкая частота возникновения опухолей у бестимусных животных и резкое повышение частоты опухолей у животных с подавленным врожденным противоопухолевым иммунитетом свидетельствуют о высокой эффективности системы иммунного надзора. В классическом варианте развития злокачественного новообразования можно условно выделить два этапа. На первом этапе происходит формирование первичного опухолевого узла, образованию которого предшествует состояние гиперплазии, метаплазии и дисплазии ткани [7]. Злокачественно трансформированная клетка, дающая начало опухолевому клону первичного опухолевого узла, возникает из пластов генетически и фенотипически измененных клеток [42]. Долгое время злокачественно трансформированные клетки первичного узла не выходят из-под регулирующего влияния микроокружения и не могут развиваться полностью автономно, хотя уже обладают устойчивостью к действию эффекторов системы иммунного надзора. Для второго этапа развития злокачественного новообразования характерно появление трансформированных клеток, способных к опухолевой прогрессии, которая подразумевает неконтролируемое разрастание в окружающей ткани (инвазия) и высокую вероятность распространения за пределы этой ткани (метастазирование). Готовность малигнизированных клеток к опухолевой прогрессии требует времени и становится возможной в случае отбора клеточных клонов, наиболее успешно ускользающих от влияний иммунной системы. Наличие «совершенного» механизма иммунной толерантности позволяет таким клеткам преодолевать в ходе инвазии и метастазирования иммунологические барьеры организма. Закономерен вопрос: существуют ли специфические гены, продукты которых обеспечивают «совершенные» и в какой-то степени универсальные механизмы иммунной толерантности? Формирование трансформированных клеток, способных к опухолевой прогрессии, равно как и клеток, образующих первичный опухолевый узел, воз можно в условиях интенсивной митотической активности нормальных клеток. Чтобы ответить на сформулированный выше ключевой вопрос, необходимо установить, имеется ли зависимость биохимических проявлений иммунной толерантности от активности определенных онкогенов или от отсутствия активности антионкогенов и влияет ли многоклеточность опухолевой ткани на формирование иммунной толерантности. Известно, что воздействие ростовых факторов на нормальную клетку сопровождается утратой ею чувствительности к цитотоксическому действию эффекторов иммунной системы в течение всего периода действия цитокинов этого типа [9]. Молекулярный механизм транзиторной толерантности нормоцитов инициируется ростовым фактором и сводится к торможению программы апоптоза, что достигается временной гиперэкспрессией ингибиторов каспаз и шаперонов, ускоренной деградацией белка р53 и изменением активности стеллажных и якорных молекул. Так, опосредованное фосфатидилинозитол 3 киназой (PI3K) и протеинкиназой В (PKB) увеличение активности антиапоптотических белков семейства Всl 2 и актив ности р70 S6 киназы при одновременной депрессии синтеза инициаторных каспаз и активности фермента GSK 3 (glycogen synthetase kinase 3), а так же опосредованный NF ?B прирост уровня цитоплазматических ингибиторов активности каспаз IAPI и IAPII являются неотъемлемыми составляющими биологической активности ростовых факторов [3, 9]. Подавляя апоптоз, ростовые факторы позволяют клеткампредшественникам опухолевого клона беспрепятственно проходить особо отслеживаемые системой иммунного надзора этапы своего развития. В злокачественно трансформированных клетках экспрессия антиапоптотических белков носит перманентный характер и осуществляется как при воздействии ауто или паракринных ростовых факторов, так и независимо от них. Примером перманентной экспрессии антиапоптотических белков, независимой от воздействия ростовых факторов, является появление у малигнизированных клеток онкогенно измененного белка р21. Белок р21 может функционировать как киназа и активировать сигнальные потоки, проводящие в клетку митогенный и антиапоптотический сигналы. Антиапоптотическая активность активированного белка р21 опосредуется PI3K/РКВ [9]. В результате мутации белок р21 может постоянно активировать митогенный и антиапоптотический сигнальные пути, а клетка будет находиться в режиме самостимуляции антиапоптотической активности и даже в условиях отсутствия ростовых факторов проявлять устойчивость к проапоптотическому воздействию эффекторов и регуляторных молекул системы иммунного надзора. На стадии злокачественного перерождения селективным преимуществом обладают именно те трансформированные клетки, у которых в цепи регуляторных сигналов белковый продукт дефектного протоонкогена располагается выше регуляторов, опосредующих антиапоптогенное действие. Активные белковые продукты дефектных протоонкогенов проводят антиапоптогенные сигналы, способные самостоятельно инициировать механизмы иммунной толерантности и обеспечивать малигнизированным клеткам устойчивость к воздействию эффекторов иммунной системы. Избыточная экспрессия ингибиторов каспаз и обусловленная ими составляющая иммунной толерантности малигнизированных клеток может быть связана не только с дефектом ключевого протоонкогена. Так, функциональная активность белкового продукта гена с myc направлена на инициацию иммунной толерантности посредством экспрессии ингибиторов каспаз [15] и мембранных транспортеров типа Рgp [14]. Гиперэкспрессия гена с myc позволяет малигнизированным клеткам преодолевать проапоптогенные и антипролиферативные эффекты антионкогенов [8]. Помимо протоонкогенов в клетках функционирует система антионкогенов, дефекты которых также могут обеспечивать иммунологическую толерантность малигнизированных клеток. Белок р53 является ключевым антионкогеном в большинстве клеток, с которыми связаны сигналы о нештатных состояниях клетки и ее окружения, инициируя устранение последствий при нештатных ситуациях путем ареста клеточного цикла или запуска апоптоза [17]. Дефекты р53 позволяют клеткам обрести толерантность к цитотоксическому действию эффекторов иммунной системы на протяжении всего злокачественного перерождения и формирования клона злокачественных клеток [17]. В настоящее время известно более 300 протоонкогенов и 100 антионкогенов [8]. Вероятно, онкогенная модификация каждого из них в той или иной степени позволяет трансформированным клеткам быть толерантными к воздействиям системы иммунного надзора, что заложено в природе трансформированной клетки. Однако каким образом одна клетка может концентрировать в геноме дефекты, рассеянные по гетерогенной популяции перерождающихся клеток Некоторый свет на эту проблему проливают результаты исследований лаборатории Г.И. Дейчмана в Институте канцерогенеза Российского онкологического научного центра им. Н.Н. Блохина РАМН. Установлено, что способность к инвазии и метастазированию злокачественно перерожденных клеток первичного опухолевого узла коррелирует с их способностью к секреции PGE2 и высокой катаболизирующей активности перекиси водорода ([Н2О2 СА+PGE2] фенотип клеток). Показано также, что малигнизированным клеткам, которые обладают трансформирующими генами myc, p53, Ras, требуется несколько циклов отбора in vivo. Клетки, трансформированные генами myc, p53 или Ras, способны образовывать первичный опухолевый узел, но не растут инвазивно и не метастазируют. Воздействие на подобные клетки перекиси и резидентных макрофагов в условиях in vitro не приводит к формированию [Н2О2 СА +PGE2] фенотипа [6]. Способность к инвазии и метастазированию появляется только у клеток, обладающих [Н2О2 СА +PGE2] фенотипом. Однако наличие варианта одноступенчатого v srk индуцированного канцерогенеза ведет к появлению [Н2О2 СА +PGE2] фенотипа клеток, которые оказываются способными к инвазии и метастазированию, и указывает, что белковые продукты srk генов являются структурными носителями иммунной толерантности. Известно, что [Н2О2 СА +PGE2] клеточным фенотипом обладают активированные макрофаги [11]. Но каким образом малигнизированные клетки заимствуют у макрофагов данный метаболический фенотип Очевидно, что заимствование этого качества происходит в структуре опухолевой ткани. Не исключается также способность злокачественно трансформированных клеток к заимствованию тех или иных генов как друг у друга, так и у нормоцитов.
Результаты изучения явления горизонтального переноса генов указывают на подобную возможность [33, 34]. Функцию вероятного переносчика структурно выполняют не столько апоптозные тельца, сколько белок острофазного ответа—сывороточный амилоид Р (SAP—serum amyloid P component). Белок SAP способен к связыванию с ДНК. При этом взаимодействии вытесняется гистон Н1. Комплекс SAP?ДНК солюбилизирует ДНК и защищает ее от действия ДНКаз [20, 21]. SAP связывается также с апоптотическими тельцами [21]. Тот факт, что в очаге опухолевого роста как нормоциты, так и малигнизированные клетки гибнут преимущественно путем некробиоза и апоптоза, повышает вероятность горизонтального переноса и формирования наиболее злокачественного и резистентного к иммунным воздействиям клона. Заключение Возникновение злокачественно трансформированной клетки и последующее развитие клона в опухоль являются фазами сложного морфогенетического процесса, интегрированного в систему жизнеобеспечения организма и претерпевающую существенную трансформацию по ходу опухолевой прогрессии. Постоянная нагрузка канцерогенами и вирусами, а также условия хронического воспалительного процесса инициируют генетическую нестабильность нормоцитов и обеспечивают вероятность появления трансформированных клеток, нейтрализующих механизмы их распознавания макрофагами и NK клетками. Генетические механизмы иммунной толерантности сопряжены с дефектами антионкогенов и горизонтальным обменом генетической информацией. Малая вероятность формирования необходимых составляющих иммунной резистентности в отдельной клетке свидетельствует в пользу того, что в процессе канцерогенеза каким то образом реализуется переход количества клеток с различающимися возможностями в новое качество. Появляется клетка, сочетающая в себе различные возможности противодействия эффекторам врожденного противоопухолевого иммунитета. Эта клетка и становится прародителем опухолевого клона. На цитоплазматическом уровне иммунная толерантность опухолевых клеток обеспечивается механизмами избирательного протеолиза. Благодаря активности протеолитических ферментов в малигнизированных клетках подавляются антипролиферативный и цитотоксический компоненты действия регуляторных и эффекторных молекул иммунной системы. Трансформированные, но не отвечающие новым условиям, клетки обречены на гибель и в свою очередь провоцируют развитие в зоне опухоли воспалительной реакции извращенного типа. Воспалительный очаг возникает и поддерживается в активном состоянии за счет функциональной активности «успешных» трансформированных клеток, которые секретируют тканеспецифичные и неспецифичные факторы, аналогичные факторам пролиферирующих и дифференцирующихся нормоцитов. Из всего спектра цитокинов и хемокинов, секретируемых опухолью и обеспечивающих ее интеграцию в систему общего жизнеобеспечения организма, ключевая роль принадлежит цитокинам с системными эффектами — IL-1 и TNF?. Как минимум, следующие четыре составляющие биологической активности данных и других цитокинов наиболее значимы для усиления биологического потенциала злокачественной опухоли и поддержания ее роста и развития. Наиболее значимы такие эффекты этих цитокинов, как стимулирование пролиферации опухолевых клеток, образование de novo сосудов в зоне опухолевого роста, индукция синтеза эндокринными железами пептидных и стероидных гормонов с последующей мобилизацией метаболических ресурсов организма, модуляция функциональной активности иммунокомпетентных клеток в структуре и микроокружении опухоли, а также изменение их способности к миграции. Таким образом, «комфортное» положение раковых клеток в иммунологически агрессивной среде достигается за счет использования ими всего комплекса механизмов обеспечения иммунной резистентности. При этом малигнизированные клетки не создают принципиально новых механизмов реализации иммунной толерантности и иммунодепрессивного воздействия на организм носителя опухоли, а используют все многообразие существующих. Вектор прогрессии самих биологически трансформированных клеток направлен в сторону отбора тех клеточных форм, которые способны автономно существовать в организме и использовать все многообразие механизмов межклеточных взаимодействий, в том числе метаболические отношения, возможности ремоделирования тканевых структур и интегративные функции нейроиммуноэндокринной системы.
ЛИТЕРАТУРА
1. Абелев Г.И. Дифференцировочные антигены в опухолях — зависимость от механизмов канцерогенеза и прогрессии // Мол. биол. — 2003. — Т. 37, № 1. — С. 4–11.
2. Антонов В.Г., Козлов В.К. Патогенез онкологических заболеваний: иммунные и биохимические феномены и механизмы. Внеклеточные и клеточные механизмы общей иммунодепрессии и иммунной резистентности // Цитокины и воспаление. — 2004. — Т. 3, № 1. — С. 8–19.
3. Белецкий И.П., Мошникова А.Б., Прусакова О.В. Пути передачи цитотоксического сигнала рецепторами семейства TNF Rs // Биохимия. — 2002. — Т. 67, вып. 4. — С. 377–395.
4. Гусев Н.Б. Протеинкиназы: строение, классификация, свойства и биологическая роль // Соросовский образовательный журнал. — 2000. — Т. 6, № 12. — С. 4–12.
5. Дас Д.К., Молик Н. Превращение сигнала гибели в сигнал выживания при редоксигнализации // Биохимия. — 2004. — Т. 69, вып. 1. — С. 16–24.
6. Дейчман Г.И. Естественный отбор и ранние изменения фенотипа опухолевых клеток in vivo: приобретение новых механизмов защиты // Биохимия. — 2000. — Т. 65, вып. 1. — С. 92–111.
7. Заридзе Д.Г. Эпидемиология и этиология злокачественных новообразований // Канцерогенез / Под ред. Д.Г. Заридзе. — М.: Научный мир. — 2000. — С. 21–56.
8. Копнин Б.П. Мишени действия онкогенов и опухолевых супрессоров // Биохимия. — 2000. — Т. 65. — С. 5–33.
9. Красильников М.А. Сигнальные пути, регулируемые фосфатидилинозит киназой и их значение для роста и выживаемости и злокачественной трансформации клеток // Биохимия. — 2000. — Т. 65, вып. 1. — С. 68–78.
10. Моргулис Б.А., Гужова И.В. Белки стресса в эукариотической клетке // Цитология. — 2000. — T. 42. — С. 323–342.
11.Ройт А., Бростофф Дж., Мейл Д. Иммунология. — М.: Мир, 2000. — С. 376–392.
12. Свердлов Е.Д. Некоторые принципы организации сигнальных систем клетки: геном — инструктор или исполнитель // Вестник РАМН. — 2001. — № 10. — С. 8–19.
16. Фильченков А.А. Каспазы: регуляторы апоптоза и других клеточных функций // Биохимия. — 2003. — Т. 68. — С. 453–456.
17. Чумаков П.М. Функция гена р53: выбор между жизнью и смертью // Биохимия. — 2000. — Т. 65. — С. 34–47.
18.Ambrosini G., Adida C., Altiery D. A novel antiapoptosis gene, survivin, expressed in cancer and lymphoma // Nat. Med. — 1997. — Vol. 3, № 9. — Р. 917–921.
19. Beere H.M., Wolf В.В., Cain K. et al. Heat shock protein 70 inhibits apoptosis by preventing recruitment of procaspase to the Apaf1 apoptosome // Nat. Cell Biol. — 2000. — Vol. 2, № 3. — P. 469–475.
20. Bickerstaff M.C., Botto M., Hutchinson W.L. at al. Serum amyloid P component controls chromatin degradation and prevents antinuclear autoimmunity // Nature Med. — 1999. — Vol. 5, № 4. — P. 694–697.
21.Butler P.J., Tennet G.A., Pepys M.B. Pentaxinchromatin interactions: serum amyloid P component specificaly displaces H1type histones and solubIL-ises native long chromatin // J. Exp. Med. — 1990. — Vol. 172, № 1. — P. 13–18.
22. Casio P., HIL-ton C., Kisselev A.F. et al. 26S proteasomes and immunoproteasome sproducemainly Nextended versions of an antigenic peptide // EMBO J. — 2001. — Vol. 20. — P. 2357–2366.
23. Chang F., Li R., Ladisch St. Shedding of gangliosides by human medulloblastoma cells // Exp. Cell. Res. — 1997. — Vol. 234, № 2. — Р. 341–346.
24. Chen F., Chang D., Goh M. et al. Role p53 in cell cycle regulation and apoptosis following exposure to proteasome inhibitors // Cell Growth Differ. — 2000. — Vol. 11, № 2. — P. 239–246.
25. Deveraux Q., Takahashi R., Salvesen GS. et al. Xlinked IAP is a direct inhibitor of cell death proteases // Nature. — 1997. — Vol. 388, № 4. — Р. 300–303.
26. Fischer U., Janicke R.U., Schulze Osthoff K. Many cuts toruin: a comprehensive update of caspase substrates // Cell Death Differ. — 2003. — Vol. 10, № 1. — Р. 71–100.
27. Finkel T., Holbrook N.J. Oxidants, oxidative stress and the biology of ageing // Nature. — 2000. — Vol. 408, № 4. — P. 239–247.
28. Garrido C., Gurbiixani S., Ravagnan L. et al. Heat shock proteins: endogenous modulators of apoptotic cell death // Biochem. Biophys. Res. Commun. — 2001. — Vol. 286, № 3. — P. 433–442.
29. Griffin T.A., Nandi D., Crus M. et al. Immunoproteasome assembly: cooperative incorporation of interferon ? (IFN?) induscible subunits // J. Exp. Med. — 1998. — Vol. 187, № 1. — P. 97–104.
30. Groll M., Ditsel L., Lowe J. et al. Structure of 20S proteasome from yeast at 2.4 A resolution // Nature. — 1997. — Vol. 386, № 6. — P. 463–471.
31.Hanahan D., Weinberg R.A. The hallmarks of cancer // Cell. — 2000. — Vol. 100, № 1. — P. 57–70.
32. He H., Qi X.M., Grossmann J., Distelhorst C.W. c Fos degradation by the proteasome. An early, Bcl 2 regulated step in apoptosis // J. Biol. Chem. — 1998. — Vol. 273, № 39. — P. 25015–25019.
33. Holcik M. The IAP proteins // Trends Genet. — 2002. — Vol. 18, № 10. — P. 537–542.
34. Holmgren L., Szeles A., Rajnavolgyi E. at al. Horisontal transfer of DNA by the apoptotic bodies // Blood. — 1999. — Vol. 93, № 7. — P. 3956–3963.
35. Homem de Bittencourt P.I., Curi R.,WIL-liams J.F. Glutathione metabolism and glutathione Sconjugate export ATPase (MRP1/GSX pump) activity in cancer. I.Differential expression in human cancer cell lines // Biochemistry and Molecular Biology International. — 1998. — Vol. 45, № 6. — Р. 1227–1241.
36. Jaattela M., Wissing D., Kokholm K. et al. Hsp70 exerts its anti-apoptotic function downstream of caspaselike proteases // Ibid. — 1998. — Vol. 17. — P. 6124–6134.
38. King R.W., Dechais R.J., Peters J.M., Kirschner M.W. How The proteolysis driwes the cell cycle // Science. — 1996. — Vol. 274, № 8. — P. 1652–1659.
39. Lee D.H., Goldberg A.L. Proteasome inhibitors: valueble new tools for cell biologists // Trends Cell Biol. — 1998. — Vol. 8, № 3. — P. 397–403.
40. Li P., Nijhawan D., Budihardjop I. et al. Cytochrome с and dATPdependent formation of Apafl/caspase 9 complex ini-tiates an apoptotic protease cascade // Cell. — 1997. — Vol. 91, № 3. — P. 479–489.
41. Loeffler M., Daugas E., Susin S. A. et al. Dominant cell death induction by extra mitochondrially targeted apoptosis inducing factor // FASEB J. — 2001. — Vol. 15, № 8. — P. 758–767.
42. Loeb L.A. A mutator phenotype in cancer // Cancer Res. — 2001. — Vol. 61. — P. 3230–3239. 43. Lopes U.G., Erhardt P., Yao R., Cooper G.M. p53 dependent induction of apoptosis by proteasome inhibitors // J. Biol. Chem. — 1997. — Vol. 272, № 9. — P. 12893–12896.
44. Lowe J., Stock D., Jap B. et al. Crystal structure of the 20S proteasome from the archaeon T. acidophum at 3.4 A resolution // Science. — 1995. — Vol. 268, № 5210. — P. 533–539.
45. Mao Y., Desai S.D., Ting C.Y. et al. 26S proteasome degradation of topoisomerase Iicleavable complex // J. Biol. Chem. — 2001. — Vol. 276, № 10.— P. 40652–40658.
46. Marso I. The permeabIL-ity transition pore complex // J. Exp. Med. — 1998. — Vol. 187, № 5. — P. 1261–1271.
47. Mitsui A., Sharp P.A. Ubiquitination of RNA polymerase II large subunit signaled by phosphrylation of carboxylterminal domain // Proc. Natl. Acad. Sci. USA. — 1999. — Vol. 96. — P. 6054–6059.
48. Mulders TMT, Keizer H.J., Breimer D.D., Mulder G.J. In vivo characterization and modulation of the glutathione/glutathione S transferase system in cancer patients// Drug Metab. Revs. — 1995. — Vol. 27. — P. 191–229.
49. Nicholson D.W. Caspase structure, proteolytic substrates, and function during apoptotic cell death // Cell Death Differ. — 1999. — Vol. 6, № 4.— P. 1028–1042.
50. Nomoto S.,Yamashita K., Koshikava et al. Mitochodrial Dloop mutations as clonal markers in multicentric hepatocellular carcinoma and plasma // Clin. Can cer Res. — 2002. — Vol. 8, № 5. — P. 481–487.
51. Obrador E., Navarro J., Mompo J. at al. Regulation of tumor cell sensitivity to TNF induced oxidative stress and cytotoxicity: Role of glutathione // Bio Factors. — 1998. — Vol. 8. — P. 23–26.
52. Pickart C.M. Mechanisms uderlying ubiqutination // Annu. Rev. Biochem. — 2001. — Vol. 70, № 3. — P. 503–533. 53. DePinho R.A. The age of cancer // Nature. — 2000. — Vol. 408, № 3. — P. 248–254.
54. Reed J.C. Dysregulation of apoptosis in cancer // J. Clin. Oncol. — 1999. — Vol. 17, № 9. — Р. 2941–2953.
55. Roy N., Deveraux Q.l., Takahashi R. et al. The c IAP 1 and c IAP
2 proteins are direct inhibitors of specific caspases // EMBO J. — 1997. — Vol. 16, № 7. — Р. 6914–6925.
56. Schall T.J., Lewis M., Koller K.J. et al. Molecular cloning and expression of a receptor for human tumor necrosis factor // Cell. — 1990. — Vol. 61, № 3. — P. 361–370.
58. Tanahashi N., Kawahara H., Murakami Y., Tanaka K. The proteasome dependent proteolytic system // Mol. Biol. Reports. — 1999. — Vol. 26, № 1. — P. 3–9.
59. Tanaka K. Molecular biology of proteasomes // Biochem. Biophys. Res. Commun. — 1998. — Vol. 247, № 4. — P. 537541.
60. Tanaka K., Kasahara M. The MHC class I ligand generating system: roles of immunoproteasomes and INF? inducible PA28 // Immunol. Rev. — 1998. — Vol. 163, № 2. — P. 161–176.
61. Thorberry N.A., Lazebnik Y. Caspases: enemies within // Science. — 1998. — Vol. 281, № 1. — Р. 12–16.
62. Waterhous N., Green D. Mitochondria and apoptosis // J. Clinical Immunol. — 1999. — Vol. 19, № 4. — P. 378–386.
63. Yang Y., Fruh K., Ahn K., Peterson P.A. In vivo assembly of the proteosomal complexes, implications for antigen processing // J. Biol. Chem. — 1995. — Vol. 270, № 10. — P. 27687–27694.
64. Yew P.R. Ubiquitinmediated proteolysys of vertebrate G1 and S phase regulators // J. Cell Physiol. — 2001. — Vol. 187, № 1. — P. 1–10.
65. Zvang H., Xu Q., Krajwski S. et al. BAR: An apoptosis regulator at the intersection of caspases and bcl 2 famIL-y proteins // Proc. Natl. Acad. Sci. USA. — 2000. — Vol. 97. — P. 2597–2602.
Pathogenesis of oncologic diseases. Cytoplasm and molecular mechanisms of immune resistance in malignant cells V.G. Antonov , V.K. Kozlov S.M. Kirov Russian Mitary Medical Academy; Medical Academy of Postgraduate Education, St. Petersburg
The role of redox reactions, heat shock proteins, proteasome complex and caspases as a molecular basis for malignant cells’ resistance to immune survelance are reviewed in the article. Mechanisms of genetic regulation of these factors, protooncogenes and antioncogenes as well as the role of cytokines are analyzed.
(Cytokines and Inflammation. 2004. Vol. 3, № 2. P. 23–33.)