Н.А. Маянский, Э. Блинк, Д. Роос, Т. Кайперс Sanquin Research at CLB, and Landsteiner Laboratory, Академический медицинский центр Амстердамского университета, Нидерланды Роль Omi/HtrA2 в каспазонезависимой клеточной гибели нейтрофилов человека
Н.А. Маянский, Э. Блинк, Д. Роос, Т. Кайперс Sanquin Research at CLB, and Landsteiner Laboratory, Академический медицинский центр Амстердамского университета, Нидерланды Роль Omi/HtrA2 в каспазонезависимой клеточной гибели нейтрофилов человека
Изучена роль митохондриальной сериновой протеазы Omi/HtrA2 в клеточной гибели нейтрофилов. В интактных клетках этот белок локализовался в межмембранном пространстве митохондрий, а при «классическом» апоптозе выделялся в цитозоль, где способствовал активации каспаз, причем специфический ингибитор сериновой протеазной активности Omi/HtrA2 — ucf 101 — не влиял на уровень спонтанного апоптоза нейтрофилов или апоптоза, вызванного воздействием только TNF? ? ? ? ?. При каспазонезависимой клеточной гибели нейтрофилов, индуцированной TNF? ? ? ? ?, протеаза Omi/HtrA2 оставалась в митохондриях, а добавление в культуру ucf 101 полностью предотвращало эту форму клеточной гибели. Приведенные результаты указывают на то, что интрамито* хондриальная активность Omi/HtrA2 как сериновой протеазы требуется для каспазонезависимой гибели нейтрофилов. (Цитокины и воспаление. 2004. Т. 3, № 2. С. 47–51.)
Omi/HtrA2 является митохондриальной сериновой протеазой, участвующей в процессе клеточной гибели [4, 10, 12]. Omi/HtrA2 млекопитающих имеет значительную гомологию с бактериальной эндопротеазой HtrA (high temperature requirement A). Последняя локализуется в периплазматическом пространстве бактерий, и ее присутствие необходимо для поддержания термотолерантности: за счет сериновой протеазной активности HtrA удаляются поврежденные и денатурированные вследствие воздействия высокой температуры или оксидантного стресса белки [11]. Роль Omi/HtrA2 в гибели клеток была обнаружена недавно, когда выяснилось, что этот белок, в норме находящийся в межмебранном пространстве митохондрий, при апоптозе выходит в цитозоль (наряду с другими митохондриальными проапоптозными факторами) [4, 10]. Здесь Omi/HtrA2 связывается с белками — ингибиторами апоптоза (inhibitor of apoptosis proteins—IAPs) и инактивирует их, высвобождая секвестрированные IAPs каспазы, которые активируются и поддерживают дальней шее развитие апоптоза (аналогично работает другой IAP регуляторный белок, выделяемый митохондриями,— Smac/DIABLO) [12]. Предполагается, что эта функция не требует протеазной активности, хотя имеются данные о том, что IAPs могут быть прямыми субстратами Omi/HtrA2 (см. ниже). Активность Omi/HtrA2 как сериновой протеазы может быть востребована при каспазонезависимой гибели клеток, хотя субстраты мишени в этом случае остаются неизвестными [4]. В настоящей работе на модели TNF? индуцированной каспазонезависимой гибели нейтрофилов [2, 5, 6] демонстрируется важная роль протеазной активности Omi/HtrA2 для данного типа гибели клеток.
Материалы и методы
Нейтрофилы выделяли и культивировали согласно описанной ранее методике [1, 6, 7]. В 24 луночный планшет помещали 1 мл (5 ? 106 клеток/мл) взвеси нейтрофилов и инкубироL вали в присутствии TNF? (Calbiochem, Germany), zVADLfmk (Alexis Biochemicals, USA), ucf 101 [3] (предоставлен A. Zervos, University of Central Florida, Orlando, USA), циклогексимида (Calbiochem) в указанных в тексте комбинациях и концентрациях (см. также подписи к рисункам). Оценку клеточной гибели проводили путем проточной цитометрии нейтрофилов, меченных аннексином VLФИТЦ и йодидом пропидия (ИП; Bender Medsystems, Austria), а также при помощи окрашивания митохондрий флуоресцентным красителем MitoTracker GreenFM (Molecular Probes, USA) с последующим подсчетом процента клеток с агрегированными митохондриями (см. рис. 2, б) [1, 6, 7]. Субклеточное фракционирование и иммуноблоттинг (Western blotting, WB) проводили следующим образом [7, 8]. Нейтрофилы отмывали в ледяном PBS (забуференный фосфатом физиологический раствор) и взвешивали в ледяном буфере для экстракции цитозоля [состав: 250 мМ сахарозы, 70 мМ KCl, 250 мкг/мл дигитонина, 1 таблетка Complete Mini protease inhibitor cockta (Roche, Germany); 2 mM диизопропилфторофосфата (DFP; Acros Organics, USA), 5 мМ ЭДТА в 5 мл PBS] в конечной концентрации 100 ? 106 клеток/мл. После 10–15 мин инкубации на льду, когда 80–90 % клеток становились проницаемыми для трипанового синего, суспензии осаждали центрифугированием (1000 ? g, 5 мин) и собирали надосадок как цитозольные фракции. Остававшийся в пробирках осадок ресуспендировали в ледяном митохондриальном лизис буфере (состав: 100 мМ NaCl, 10 мМ MgCl2 ?6H2O, 2 мМ ЭДТА, 2 мМ ЭГТА, 1 % детергента NPL40, 10 % глицерина, 1 таблетка коктейля ингибиторов протеаз, 2 мМ DFP в 5 мл 50 мМ Tris буфера, рН 7,5) в объеме, равном объему буфера, использованного для экстракции цитоL золя. После 10 минутной инкубации на льду проводили центрифугирование (10 000 ? g, 10 мин) и полученный надосадок собирали как митохондриальные фракции. Для приготовления образцов для WB 24 мкл цитозольной/митохондриальной фракции смешивали с 8 мкл четырехкратного загрузочного буфера, содержащего додецилсульфат натрия (sodium dedecyl sulfate sample buffer, SDSLSB; состав 4 ? SDSLSB: 5 % SDS, 40 % глицерина, ~0,01 % красителя Comassie blue в 125 мМ Tris буфере, рН 6,8) и 8 % ? меркаптоэтанола, и кипятили в течение 5 мин, после чего полученные образцы использовали для электрофореза. Электрофорез белков проводили в 12,5% ом полиакриламидном геле, содержащем SDS (SDSLPAGE). Для WB использовали поликлональные антитела против Apaf1 (Pharmingen, USA) и MnСОД (Stressgen, Canada) в разведении 1 : 1000. Анти Omi/HtrA2 поликлональные антитела [4] были предоставлены проф. E.S. Alnemri (Thomas Jefferson University, Phadelphia, USA) и использовались в разведении 1 : 5000. Внутриклеточную локализацию ucf 101 определяли с помощью конфокального микроскопа (LSM510, Carl Zeiss, Germany). Нефиксированные нейтрофилы инкубировали с 20 мкМ ucf 101 и 100 нМ MitoTracker GreenFM в течение 15 мин при 37 °С, отмывали и микроскопировали согласно [7]. Анализ изображения проводили с использованием пакета программ LSM510 Image software (Carl Zeiss).
Результаты и обсуждение
При изучении механизмов каспзазонезависимой клеточной гибели нейтрофилов, индуцированной TNF? [2, 6], мы сделали следующее наблюдение. Нейтрофилы живут недолго, быстро апоптируя при культивировании in vitro без дополнительных воздействий [9]. Уже после 6 ч инкубации 20–25 % клеток подвергались спонтанному апоптозу и связывали аннексин V (рис. 1, а). Добавление в культуру только TNF? вызывало «классический» каспазозависимый апоптоз [2, 6]. Однако в присутствии ингибитора каспаз zVAD fmk TNF? парадоксально усиливал повреждение клеток, индуцируя «неклассическую» каспазонезависимую гибель нейтрофилов, когда более 50 % клеток в течение 6 ч становились аннекcин V+ (рис. 1, б) [2, 6]. Клеточную гибель, вызванную комбинацией TNF? zVAD fmk, удалось предотвратить внесением в инкубационную стреду ucf 101— специфического ингибитора сериновой протеазной активности Omi/HtrA2 [3], который значительно снижал количество аннексин V+ нейтрофилов в этих условиях (рис. 1, в). Количество аннексин V+клеток коррелировало с числом клеток, имевших изменения в митохондриях, которые оценивались при помощи специфического флуоресцентного красителя митохондрий MitoTracker GreenFM [6, 7]. Практически все свежевыделенные нейтрофилы имели трубчатые митохондрии (рис. 2, а), которые трансформировались в апоптировавших
Рис. 1. Клеточная гибель нейтрофилов, по данным окрашивания аннексином LV и йодидом пропидия (ИП)
Нейтрофилы человека, выделенные из периферической крови здоровых доноров, инкубировали в течение 6 (а, в) или 20 (г, д) ч без добавлений (а, г), с комбинацией 50 нг/мл TNF? и 150 мкмоль/л zVADLfmk (б) или с TNF?+zVADLfmk в присутствии 20 мкмоль/л ucf 101 (в) или только с ucf 101 (д). После культивирования клетки окрашивали аннексиномLVLФИТЦ и ИП и анализировали на проточном цитометре. Цифры указывают процент аннексин V+ Lклеток (нижний + верхний правые квадранты) в обозначенных культурах по результатам 4–6 независимых экспериментов (M ± m).
Рис. 2. Митохондрии в нейтрофилах
Свежевыделенные (а) и спонтанно апоптировавшие после 20 часовой инкубации (б) нейтрофилы окрашивали MitoTracker GreenFM (специфическим для митохондрий флуоресцентным красителем) и анализировали при помощи флуоресцентного микроскопа. В контрольных нейтрофилах митохондрии имели трубчатую структуру, а в апоптозных клетках они превращались в округлые агрегаты (отрезок — 5 мкм).
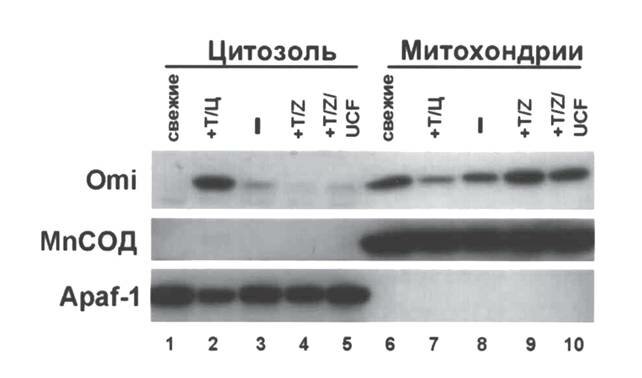
клетках в округлые агрегаты (рис. 2, б), причем число клеток с митохондриальными кластерами примерно совпадало с числом апоптозных нейтрофилов [6]. Агрегированные митохондрии имели 19 ± 2 % клеток из культур без добавлений, 51 ± 10 % клеток, обработанных TNF? + zVAD fmk, и 20 ± 6 % клеток, инкубированных с TNF? + zVAD fmk + ucf 101 (M ± m; n = 3–4). Очевидно, что ucf 101 препятствовал деградации митохондрий, индуцированной комбинацией TNF? + zVAD fmk. Таким образом, Omi/HtrA2 наряду с ролью в апоптозе ряда клеточных линий [4, 10] играет роль и в клеточной гибели первичных клеток (нейтрофилов). Вместе с тем, ucf 101 (в концентрации от 20 до 100 мкМ) не задерживал ни спонтанный апоптоз нейтрофилов в 20 часовых культурах (рис. 1, г, д), ни апоптоз, вызванный воздействием TNF? (данные не представлены). Следовательно, активность Omi/HtrA2 как сериновой протеазы (TNF? + zVAD fmk индуцированная клеточная гибель) не играет важной роли в процессе «классического» каспазозависимого апоптоза. Приведенные наблюдения подчеркивают двойственную природу апоптогенности Omi/HtrA2, которая, с одной стороны, обусловлена связыванием и инактивацией IAPs [4, 12], а с другой—протеолитической активностью [3]. Omi/HtrA2 опосредованная инактивация IAPs, вероятно, достаточна для развития апоптоза нейтрофилов по обычному пути (например, старение в 20 часовой культуре), а протеазная активность Omi/HtrA2 в данном случае не требуется (см. рис. 1, г, д). Однако, по данным, полученным S.M. Srinivasula и др. [13], при трансфекции клеточной линии 293, мутированной формой Omi/HtrA2, не имевшей протеазной активности, активность каспаз и клеточная гибель заметно снижались, тогда как трансфекция неизмененным, активным белком индуцировала обе реакции. Это сопровождалось деградацией ХIAP белка (один из представителей IAPs) в трансфицированных клетках, который оказался субстратом Omi/HtrA2 [13, 15]. Следовательно, механизм участия Omi/HtrA2 в клеточной гибели может варьировать в зависимости от типа клеток. С целью дальнейшего изучения роли Omi/HtrA2 в клеточной гибели нейтрофилов было исследовано перераспределение этого белка в субклеточных фракциях нейтрофилов, культивированных в различных условиях. Обычно Omi/HtrA2 локализуется в митохондриях, перемещаясь при апоптозе в цитозоль [4, 10]. Это оказалось справедливым и для нейтрофилов, поскольку в свежевыделенных клетках белок Omi/HtrA2 присутствовал в митохондриях (рис. 3, дор. 6). При индукции апоптоза, например, путем инкубации нейтрофилов в течение 3 ч с комбинацией TNF? + циклогексимид, когда 70–80 % клеток становились аннексин V+ [8], Omi/HtrA2 выделялся в цитоплазму (рис. 3, дор. 2) с реципрокным снижением сигнала в митохондриальной фракции (рис. 3, дор. 7).
Выход Omi/HtrA2 в цитозоль наблюдался также и в нейтрофилах, апоптировавших спонтанно в суточных культурах (данные не показаны). Несмотря на выраженную гибель клеток после 6 часовой инкубации с TNF? + zVAD fmk (см. рис. 1, б), Omi/HtrA2 оставался в митохондриях (рис. 3, дор. 4 и 9). В присутствии ингибитора ucf 101, отменявшего TNF? + zVAD fmk зависимую клеточную гибель, Omi/HtrA2 также не покидал митохондрии (рис. 3, дор. 5 и 10). Чистота субклеточных фракций нейтрофилов была подтверждена при помощи определения экспрессии цитоплазматического белка Apaf 1 (рис. 3, дор. 1–5) и митохондри ального белка MnСОД (марганец зависимой митохондриальной изоформы фермента супероксиддисмутазы; рис. 3, дор. 6–10).
Рис. 3. Субклеточное фракционирование нейтрофилов, инкубированных в различных условиях
Нейтрофилы: свежевыделенные (дор. 1 и 6), после 3 часовой инкубации с TNF? (50 нг/мл) и циклогексимидом (2 мкг/мл) (индукция апоптоза; Т/Ц, дор. 2 и 7), после 6 часовой инкубации без добавлений (–, дор. 3 и 8), с добавлением TNF? + zVADLfmk (Т/Z, дор. 4 и 9) или TNF? + zVADLfmk и ucf 101 (T/Z + ucf, дор. 5 и 10) — фракционировали при помощи дигитонина. После этого цитозольные (дор. 1–5) и митохондриальные (дор. 6–10) фракции подвергали электрофорезу в полиакриламидном геле. Для WB применяли специфические антитела к указанным белкам. Каждая фракция нейтрофилов представляет ~2?106 клеток. Антитела к MnСОД и Apaf 1 использовали для контроля чистоты митохондриальных и цитозольных фракций соответственно. Репрезентативный результат одного из трех независимых экспериментов.
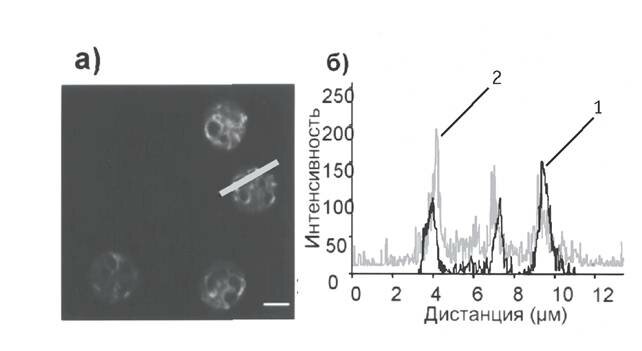
Отсутствие субклеточного перераспределения Omi/HtrA2 при TNF? + zVAD fmk индуцированной гибели нейтрофилов вместе с выявленной способностью ucf 101 (специфического ингибитора Omi/HtrA2) полностью блокировать этот механизм клеточной гибели позволяют заключить, что при определенных условиях Omi/HtrA2 может поддерживать клеточную гибель через свою протеазную активность, находясь внутри митохондрий. Действительно, при исследовании субклеточной локализации ucf 101 в нейтрофилах было обнаружено, что красная аутофлуоресценция этого ингибитора и зеленое свечение митохондриального маркера MitoTracker GreenFM совпадали (рис. 4, а; сдвиг флуресценции в желтый спектр отражает колокализацию сигналов). Колокализация была подтверждена с помощью компьютерной программы конфокального микроскопа, которая показала соответствие пиков флуоресценции ucf 101 (линия 2) пикам флуоресценции MitoTracker (линия 1) (рис. 4, б). Это свидетельствует о митохондриальной локализации ucf 101 и подтверждает, что Omi/HtrA2 ингибируется интрамитохондриально.
Рис. 4. Внутриклеточная локализация ucfL101 в нейтрофилах
Свежевыделенные нейтрофилы инкубировали с MitoTracker GreenFM (100 нМ) и ucfL101 (20 мкМ) в течение 15 мин при 37 °С, после чего определяли внутриклеточную локализацию ucf 101 с помощью конфокального микроскопа; а — колокализация MitoTracker и ucfL101 проявлялась в виде сдвига флуоресценции в желтый спектр (отрезок — 5 мкм); б — колокализация MitoTracker (линия 1) и ucfL101 (линия 2) в области, отмеченной на рис. 4 (а) косой чертой, показана при помощи компьютерного анализа этого микрофото (программа LSM510 Image). Результаты 4 независимых экспериментов.
Исходя из приведенных результатов можно сделать и другое интересное заключение. В нейтрофилах для повышения проницаемости митохондрий и выхода Omi/HtrA2 в цитозоль после воздействия TNF? требовалась активность кас паз, поскольку zVAD fmk (блок каспаз) предотвращал обе эти реакции (см. рис. 3, дор. 2, 7 и 4, 9), по крайней мере, в краткосрочных культурах. Вероятно, что регуляция описываемых процессов осуществляется за счет перераспределения белка Вах из цитозоля (интактные клетки) в митохондрии (апоптозные клетки) [1, 7], которое наблюдалось в нейтрофилах, инкубированных только с TNF?, но было заблокировано в клетках, обработанных TNF? + zVAD fmk [2, 6]. Слияние Вах с митохондриями наблюдалось также и в нейтрофилах после воздействия TNF? + циклогексимидом [8], использованного в настоящей работе.
Таким образом, ингибирование каспаз в присутствии TNF? задерживает выход Omi/HtrA2 из митохондрий в цитозоль, однако не влияет на передачу сигналов в системе «неклассической» клеточной гибели (см. рис. 1, б), которые по-прежнему могут стимулировать активность Omi/HtrA2 как сериновой протеазы, но в данном случае внутри митохондрий. Вероятные мишени протеазной активности Omi/HtrA2 неизвестны. Omi/HtrA2 опосредованный протеолиз может инактивировать антиапоптозные молекулы, как это было продемонстрировано на примере IAPs [13, 15], или же активировать какой-либо белок, чья функция требуется для каспазонезависимой клеточной гибели нейтрофилов. Данный тип клеточной гибели развивается при участии активных форм кислорода митохондриального происхождения [2, 6], поэтому логично предположить, что в этих условиях может разрушаться митохондриальная антиоксидантная система, которая обычно деактивирует избыток таких молекул. Возможно, Omi/HtrA2 вовлечен в выключение подобной системы, например, ингибируя MnСОД—антиоксидантный фермент митохондрий с высокой экс прессией в нейтрофилах (см. рис. 3, дор. 6–10) [8], который особенно важен для предотвращения TNF? индуцированного оксидантного повреждения клеток [14]. Таким образом, в настоящей работе показано, что в свежих нейтрофилах Omi/HtrA2 экспрессирован в митохондриях, но при апоптозе он перемещается в цитозоль. Кроме того, важно отметить, что активность Omi/HtrA2 в качестве сериновой протеазы требуется для каспазонезависимой, неклассической клеточной гибели нейтрофилов, индуцированной TNF?. После воздействия TNF? + zVAD fmk белок Omi/HtrA2 остается в митохондриях, что предполагает его способность поддерживать клеточную гибель нейтрофилов, находясь внутри митохондрий и, соответственно, атакуя интрамитохондриальные субстраты, которые пока остаются невыясненными.
ЛИТЕРАТУРА
1. Маянский Н.А. Субклеточное перераспределение Вах и его слияние с митохондриями при спонтанном апоптозе нейтрофилов // Иммунология. — 2001. — № 6. — С. 29–32.
2. Маянский Н.А., Роос Д., Кайперс Т. Каспазонезависимый путь клеточной гиL бели нейтрофилов человека, индуцированный ТНФ? // Цитокины и воспаление. — 2003. — Т. 2, № 1. — C. 29–35.
3. Centi L., Lee Y., Hess S. et al. Characterization of a novel and specific inhibIL- tor for the proapoptotic protease Omi/HtrA2 // J. Biol. Chem. — 2003. — Vol. 278. — P. 11489–11494.
4. Hegde R., Srinivasula S.M., Zhang Z. et al. Identification of Omi/HtrA2 as a mtochondrial apoptotic serine protease that disrupts inhibitor of apoptosis proteincaspase interaction // J. Biol. Chem. — 2002. — Vol. 277. — P. 432–438.
5. Liu C.Y., Takemasa A., Les W.C. et al. Broadspectrum caspase inhibition paradoxically augments cell death in TNFLalpha stimulated neutrophs // Blood. — 2003. — Vol. 101. — P. 295–304.
6. Maianski N.A., Roos D., Kuijpers T.W. Tumor necrosis factor a induces a caspasendependent death pathway in human neutrophIL-s // Blood. — 2003. — Vol. 101. — P. 1987–1995.
7. Maianski N.A., Mul F.P.J., van Buul J.D. et al. Granulocyte colonyLstimulating factor inhibits the mitochondriadependent activation of caspase in neuL trophIL-s // Blood. — 2002. — Vol. 99. — P. 672–679.
8. Maianski N.A., Geissler J., Kuijpers T.W., Roos D. Functional characterization of mitochondria in neutrophs: a role restricted to apoptosis // Cell Death Differ. — 2004. — Vol. 11. — P. 143–153.
9. Maianski N.A., Maianski A.N., Kuijpers T.W., Roos D. Apoptosis of neutrophs // Acta Haematol. — 2004. — Vol. 111. — P. 56–66.
10. Newmeyer D.D., FergusonLMIL-ler S. Mitochondria: releasing power for life and unL leashing the machineries of death // Cell. — 2003. — Vol. 112. — P. 481–490.
11. Pallen M.J., Wren B.W. The HtrA famy of serine proteases // Mol. Microbiol. — 1997. — Vol. 26. — P. 209–221.
12. Salvesen G.S., Duckett C.S. IAP proteins: blocking the road to death’s door // Nat. Rev. Mol. Cell Biol. — 2002. — Vol. 3. — P. 401–410.
13.Srinivasula S.M., Gupta S., Datta P. et al. Inhibitor of apoptosis proteins are substrates for the mitochondrial serine protease Omi/HtrA2 // J. Biol. Chem. — 2003. — Vol. 278. — P. 31469–31472.
14. Wong G.H., Elwell J.H., Oberley L.W., Goeddel D.V. Manganous superoxide disL mutase is essential for cellular resistance to cytotoxicity of tumor necrosis facL tor // Cell. — 1989. — Vol. 58. — P. 923–931.
15. Yang QLH., ChurchLHajduk R., Ren J. et al. Omi/HtrA2 catalytic cleavage of inL hibitor of apoptosis (IAP) irreversibly inactivates IAPs and facitates caspase activity in apoptosis // Genes Dev. — 2003. — Vol. 17. — P. 1487–1496.
Role of Omi/HtrA2 in a caspaseindependent cell death of human neutrophs N.A. Maianski, E. Blink, D. Roos and T. Kuijpers Sanquin Research at CLB, and Landsteiner Laboratory, Academic Medical Centre, University of Amsterdam, Amsterdam, The Netherlands
Omi/HtrA2 is a mitochondrial serine protease that has a proapoptotic role in mammalian cells. In present study we investigated the role of this protein in neutrophcell death. Omi/HtrA2 was expressed in the mitochondria of intact neutrophs and was normally released into the cytosol upon apoptosis. In contrast, the caspaseindependent route of neutrophcell death (induced by TNF? ? ? ? ?) proceeded without release of Omi/HtrA2. Using ucf 101, a specific inhibitor of Omi/HtrA2’s serine protease activity, we found that this activity was required for, and its inhibition protected against, this type of neutrophcell death. Our results also indicated that Omi/HtrA2 may support cell death from within the mitochondria.
(Cytokines and Inflammation. 2004. Vol. 3, № 2. P. 47–51.)